
Los estudios de asociación del genoma completo han identificado polimorfismos comunes en o cerca de TLR7 que se segregan con SLE. En ratones, el aumento de la señalización de TLR7 o a la expresión transgénica de TLR7 exaceraba la autoinmunidad y la eliminación de TLR7 previene o mejora la enfermedad en otros modelos de lupus. La mayoría de los modelos de lupus de ratón en los que TLR7 tiene un papel en la patogenicidad muestran una mayor formación de GC y células T auxiliares foliculares y se ha propuesto que TLR7 impulsa GC enriquecidas en células B autorreactivas. Sin embargo, informes recientes han demostrado que el lupus puede desarrollarse independientemente de los GC en modelos de ratones en los que la enfermedad depende de la señalización de MyD88.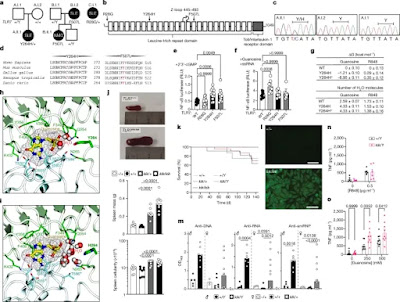

Se realizó una secuenciación del genoma de una niña española a la que se le diagnosticó con LES a los 7 años. Primero presentó trombocitopenia autoinmune refractaria y tenía anticuerpos antinucleares (ANA) elevados e hipocomplementemia. Continuó desarrollando artralgias inflamatorias, síntomas constitucionales, episodios intermitentes de hemicorea, y presentó influencia mitral leve y afectación renal tras ingreso con crisis hipertensiva. El análisis bioinformático reveló una variante sin sentido de TLR7p.Tyr264His (Y264H) que se predijo que sería dañina por SIFT y CADD. Esta variante no estaba presente en las bases de datos de variación del genoma humano normal. El residuo de tirosina mutado se encuentra en la octava repetición rica en leucina de TLR7, dentro de la parte endosomal del receptor, y está altamente conservado en todas las especies, incluido el pez cebra. Los análisis adicionales de variantes raras en 22 genes que pueden causar LES humano cuando mutan revelaron una variante heterocigota en RNASEH2B, p. Ala177Thr, que, cuando es homocigota, causa LES.
El fenotipado en profundidad de los ratones kika reveló una marcada trombocitopenia, como se ve en un recuento de glóbulos blancos ligeramente más bajo. Se evidenció glomerulonefritis proliferativa en los riñones, así como matriz mesangial expandida con depósitos electro densos y celularidad mesangial aumentada. Se observaron infiltrados linfoides en el hígado, las glándulas salivales y el páncreas, donde ocasionalmente formaron estructuras foliculares periislotes. El tejido pancreático exocrino a menudo se reemplazaba por grasa, particularmente en ratones Tlr7kik/kik. Otros hallazgos incluyeron infiltrados subpleurales, perivasculares e intersticiales en los pulmones; degeneración y necrosis de mitocitos en el músculo del panículo de la piel; fibrosis miocárdica focal, linfomas esplénicos (4 de 6 ratones); linfadenitis crónica en los ganglios linfáticos y el intestino; e hiperplasia de las placas de Peyer. Los niveles séricos de IFNy, IL-10 y TNF aumentaron.
los riñones, así como matriz mesangial expandida con depósitos electro densos y celularidad mesangial aumentada. Se observaron infiltrados linfoides en el hígado, las glándulas salivales y el páncreas, donde ocasionalmente formaron estructuras foliculares periislotes. El tejido pancreático exocrino a menudo se reemplazaba por grasa, particularmente en ratones Tlr7kik/kik. Otros hallazgos incluyeron infiltrados subpleurales, perivasculares e intersticiales en los pulmones; degeneración y necrosis de mitocitos en el músculo del panículo de la piel; fibrosis miocárdica focal, linfomas esplénicos (4 de 6 ratones); linfadenitis crónica en los ganglios linfáticos y el intestino; e hiperplasia de las placas de Peyer. Los niveles séricos de IFNy, IL-10 y TNF aumentaron.

Fuente: Nature
Comentarios
Publicar un comentario